Exercise: Day 5 - Expression Analysis with RNA-seq
Get the data
Download the following data of from https://www.dropbox.com/sh/gcsyyf4wumou1gq/AADTP8Z3Vgv7Teja1iEI6ls-a?dl=0
PRAD_raw_counts.txt - Raw counts on 86 matched tumor-normal samples and 55635 genes
PRAD_sample_info.txt - Labels associated to the samples (“Primary Tumor” and “Solid Tissue”)
Load the data into R
Do this by either first downloading the file and providing the local path:
rm(list=ls()) # clean the workspace
setwd("path_to_files") #replace location with local path_to_file
prad_data <- read.table("PRAD_raw_counts.txt",sep="\t",header=T)
RPKM values
Assuming the gene lengths defined below, write a function that, receiving in input the counts from each sample and the gene lengths, calculate the RPKM values
head(prad_data[,1:5])
## TCGA_HC_8260_11A TCGA_HC_8259_11A TCGA_EJ_7123_11A TCGA_G9_6496_01A TCGA_EJ_7781_01A
## TSPAN6 3829 3990 2770 2666 4454
## TNMD 10 23 24 5 6
## DPM1 1555 2108 1987 551 1531
## SCYL3 1096 1598 1477 426 1792
## C1orf112 236 279 307 75 273
## FGR 222 382 765 203 338
gene_lengths<-rep(c(100,1000),dim(prad_data)[1])[dim(prad_data)[1]]
returnRPKM <- function(counts, gene_lengths) {
....
return(rpkm)
}
prad_data_rpkm<-apply(prad_data,2,function(x) returnRPKM(x,gene_lengths))
head(prad_data_rpkm[,1:5])
## TCGA_HC_8260_11A TCGA_HC_8259_11A TCGA_EJ_7123_11A TCGA_G9_6496_01A TCGA_EJ_7781_01A
## TSPAN6 689.840938 602.398597 233.542303 638.641255 666.9722899
## TNMD 1.801622 3.472473 2.023471 1.197752 0.8984809
## DPM1 280.152170 318.259710 167.526555 131.992247 229.2623655
## SCYL3 197.457735 241.261393 124.527791 102.048453 268.3462828
## C1orf112 42.518271 42.122609 25.883569 17.966277 40.8808790
## FGR 39.996001 57.673249 64.498145 48.628723 50.6144216
Data handling and diagnostic plots
A. Starting from the raw count data, filter out the genes characterized by a maximum expression level across the samples which is not above 10 and log2 transform the data, using an offset =1
....
dim(prad_data_filt)
## [1] 29741 86
B. Perform the PCA analysis and plot the first two components, highlighting Primary Tumor vs Solid Tissue Normal classification (NOTE: use pch=19 and col=condition)
samp_info<-read.table("PRAD_sample_info.txt",sep="\t",header=T)
condition<-as.factor(samp_info[,2])
....

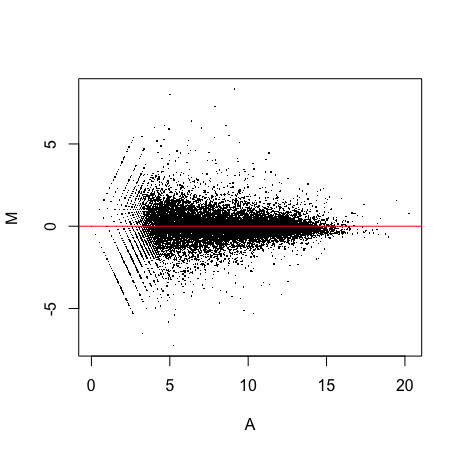
C. Write a function able to display, for a sample given as input, its MA plot versus the “median sample”, i.e. the sample of the median expression values across the genes (NOTE: use the parameter pch=”.” in the plot, and abline to highlight the 0 line)
MAplot<-function(data,n) #n = number indicating the sample
{
....
}
MAplot(prad_data_filt,n=1)
D. Install and load the package “arrayQualityMetrics”, follow the instructions below and look at the resulting report.
source("https://bioconductor.org/biocLite.R")
biocLite("arrayQualityMetrics")
library(arrayQualityMetrics)
condition<-samp_info[,2]
ggt<-data.frame(Group=condition)
row.names(ggt)<-samp_info[,1]
lab<-new("AnnotatedDataFrame",data=ggt)
ddt<-new("ExpressionSet",exprs=as.matrix(prad_data_filt),phenoData=lab)
datx<-prepdata(ddt,intgroup="Group",do.logtransform=FALSE)
arrayQualityMetrics(ddt,outdir="PRAD_raw_counts",intgroup="Group")
Normalization with DESeq2
Install and load the library DESeq2 and use the functions “DESeqDataSetFromMatrix”,”estimateSizeFactors”” and “counts” to obtain the normalized count, starting from the filtered raw count data, NOT log2 transformed. Generate the QC report (using the log2 transformed data plus offset=1) for these data and look how the dignostic plots change with respect to the report obtained from the raw data.
source("https://bioconductor.org/biocLite.R")
biocLite("DESeq2")
library("DESeq2")
ll<-apply(prad_data,1,max)
ind<-which(ll>10)
prad_data_filt<-prad_data[ind,]
condition<-as.factor(samp_info[,2])
ddsMat <- DESeqDataSetFromMatrix(countData = prad_data_filt,DataFrame(condition), ~ condition)
.... #create matnorm
head(matnorm[,1:5])
## TCGA_HC_8260_11A TCGA_HC_8259_11A TCGA_EJ_7123_11A TCGA_G9_6496_01A
## TSPAN6 4043.35607 3585.40222 1801.94582 5351.86640
## TNMD 10.55982 20.66773 15.61253 10.03726
## DPM1 1642.05241 1894.24258 1292.58713 1106.10592
## SCYL3 1157.35656 1435.95808 960.82093 855.17445
## C1orf112 249.21181 250.70858 199.71024 150.55888
## FGR 234.42806 343.26407 497.64930 407.51271
TCGA_EJ_7781_01A
TSPAN6 3956.467495
TNMD 5.329772
DPM1 1359.980183
SCYL3 1591.825270
C1orf112 242.504631
FGR 300.243829
condition<-as.character(samp_info[,2])
ggt<-data.frame(Group=condition)
row.names(ggt)<-as.character(samp_info[,1])
lab<-new("AnnotatedDataFrame",data=ggt)
ddt<-new("ExpressionSet",exprs=log2(matnorm+1),phenoData=lab)
datx<-prepdata(ddt,intgroup="Group",do.logtransform=FALSE)
arrayQualityMetrics(ddt,outdir="PRAD_norm_counts",intgroup="Group")
Differential analysis with DESeq2
Firstly, it is better to be sure that the “Solid Tissue Normal” is the first level in the condition factor, so that the default log2 fold changes are calculated as tumor over normal. The function relevel achieves this:
ddsMat$condition <- relevel(dds$condition, "Solid Tissue Normal")
The DESeq2 analysis can now be run with a single call to the function DESeq:
dds <- DESeq(ddsMat)
Calling “results” without any arguments will extract the estimated log2 fold changes and p values:
res <- results(dds)
As res is a DataFrame object, it carries metadata with information on the meaning of the columns:
mcols(res, use.names=TRUE)
## DataFrame with 6 rows and 2 columns
## type
## <character>
## baseMean intermediate
## log2FoldChange results
## lfcSE results
## stat results
## pvalue results
## padj results
## description
## <character>
## baseMean mean of normalized counts for all samples
## log2FoldChange log2 fold change (MAP): condition Primary Tumor vs Solid Tissue Normal
## lfcSE standard error: condition Primary Tumor vs Solid Tissue Normal
## stat Wald statistic: condition Primary Tumor vs Solid Tissue Normal
## pvalue Wald test p-value: condition Primary Tumor vs Solid Tissue Normal
## padj BH adjusted p-values
The first column, baseMean, is a just the average of the normalized count values, dividing by size factors, taken over all samples. The remaining four columns refer to a specific contrast, namely the comparison of the tumor samples over the normal samples for the factor variable condition. Look at the help page for results.
The column log2FoldChange is the effect size estimate. It tells us how much the gene’s expression seems to have changed due to treatment with dexamethasone in comparison to untreated samples. This value is reported on a logarithmic scale to base 2: for example, a log2 fold change of 1.5 means that the gene’s expression is increased by a multiplicative factor of (2^1.5)≈2.82.
This estimate has an uncertainty associated with it, which is available in the column lfcSE, the standard error estimate for the log2 fold change estimate. DESeq2 performs for each gene a hypothesis test to see whether evidence is sufficient to decide against the null hypothesis that there is no effect of the tumor on the gene and that the observed difference between tumor and control was merely caused by experimental variability (i.e., the type of variability that you can just as well expect between different samples in the same condition group). As usual in statistics, the result of this test is reported as a p value, and it is found in the column pvalue.
To look at the genes with a p value below 0.05:
sum(res$pvalue < 0.05, na.rm=TRUE)
## [1] 16044
Now, assume for a moment that the null hypothesis is true for all genes, i.e., no gene is affected by the condition tumor vs normal. Then, by the definition of p value, we expect up to 5% of the genes to have a p value below 0.05. However, we have more genes selected than the 5% of the total. DESeq2 uses the Benjamini-Hochberg (BH) adjustment as described in the base R p.adjust function in order to decrease the number of false positives.
A. Look at the number of genes with adjusted p-value below 5%.
....
## [1] 14369
To select the number of significant genes:
resSig <- subset(res, padj < 0.05)
B. Use DEseq2 function plotMA and use a threshold for alpha = 0.05

Final Exercise Download the GBM data from the same link reported above and repeat the pipeline. Use DESeq2 with the condition tumor vs. normal and try to combine this condition with the age or the gender as in the example:
ddsMat <- DESeqDataSetFromMatrix(countData = gbm_data_filt,.... ~ condition+gender)